
About the Author:
Jeff Nunn is the founder of Project Biohacking. With over 30 years of biohacking practice, he applies decades of self-experimentation methodology to peptide research, dosing math, and vendor evaluation.
A complete, data-driven guide to identifying signal peptides using amino acid patterns, computational models, and laboratory methods
A signal peptide is a short sequence of amino acids located at the N-terminus of a newly synthesized protein. Its job is specific: direct that protein to the right cellular destination. Without it, proteins cannot reach the compartments where they are needed, and the biochemical machinery of the cell breaks down.
Signal peptides are not functional proteins in their own right. They are targeting instructions encoded within the protein sequence itself, read by cellular components during and immediately after translation at the ribosome. Once the signal peptide has done its job, it is removed. What remains is a mature, functional protein in the correct location.
In the broader context of peptide biology, signal peptides represent one of the most fundamental regulatory mechanisms at the cellular level. They govern protein biosynthesis outcomes by controlling where proteins go, not just what they do. This distinction matters for any serious analysis of molecular physiology.
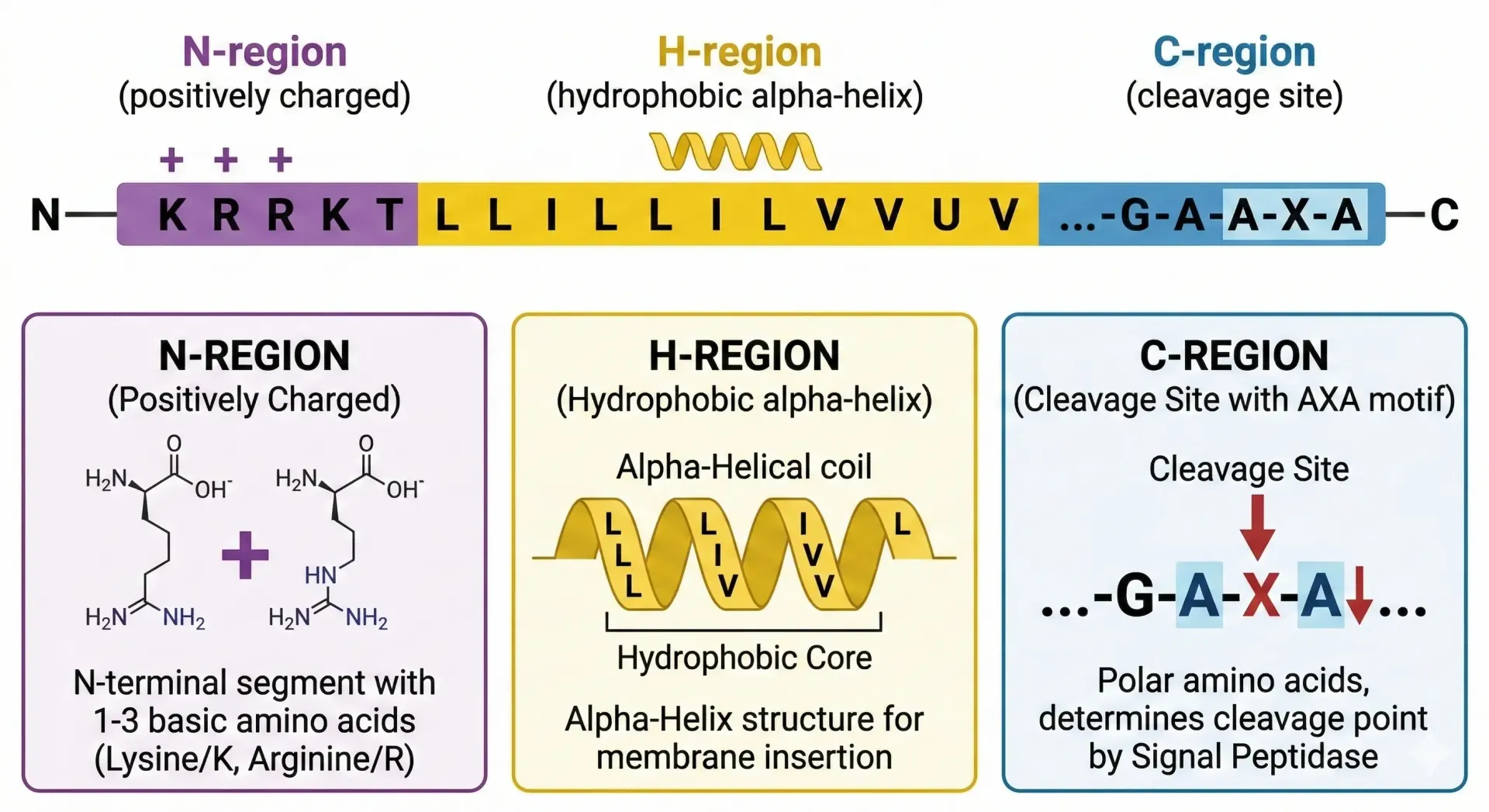
Signal peptides share a conserved three-region architecture, even though their exact amino acid sequences vary considerably between proteins and species.
The N-region sits at the far N-terminus. It carries a net positive charge, typically contributed by basic amino acids such as lysine and arginine. This positive charge plays a role in orienting the peptide relative to the membrane.
The H-region is the defining structural feature. It is a stretch of hydrophobic amino acids, typically 6 to 15 residues long, that forms a helical structure capable of inserting into a lipid bilayer. Hydrophobicity here is not incidental. It is the physical property that makes membrane interaction possible. The chemical bond geometry of this helix and the nonpolar side chains that line it create the thermodynamic conditions required for translocation.
The C-region follows the H-region and contains the cleavage site. This is where signal peptidase, an enzyme located on the luminal face of the endoplasmic reticulum membrane, cuts the signal peptide from the mature protein. The sequence surrounding the cleavage site follows a recognized pattern, often described by the AXA rule, where small neutral amino acids occupy specific positions flanking the cut site.
Understanding this structure is essential before attempting any identification. Whether working from sequence data or experimental output, the three-region model is the conceptual anchor.
The primary function of a signal peptide is to initiate co-translational translocation into the endoplasmic reticulum. As the ribosome synthesizes a protein, the emerging signal peptide is recognized by a cytoplasmic complex called the Signal Recognition Particle. This particle pauses translation and docks the ribosome to the translocon, a protein channel in the endoplasmic reticulum membrane.
Once docked, translation resumes and the growing polypeptide is threaded directly into the lumen of the endoplasmic reticulum. This process underlies the secretion pathway, the cellular mechanism by which proteins destined for the extracellular matrix, the plasma membrane, or export outside the organism are processed and transported.
Signal peptides also participate in regulation and communication at the cellular level. Their efficiency, cleavage timing, and interaction with downstream machinery influence how much of a given protein gets produced and correctly localized. Mutations in signal peptide sequences can misdirect proteins, reduce secretion efficiency, or trigger protein degradation, all of which have downstream consequences for cellular function.
Identification begins with the amino acid sequence. The methodology follows a pattern-recognition logic: look for the characteristic features of the three structural regions, starting from the N-terminus.
In practice, sequence analysis involves scanning the first 15 to 30 residues of a protein for the following indicators:
A short positively charged N-region. One to five residues with basic character at the extreme N-terminus.
A central hydrophobic stretch. A continuous run of nonpolar residues long enough to form a transmembrane helix. Interruptions in this region, or stretches that are too short, reduce the likelihood of a functional signal peptide.
A polar C-region containing a cleavage site. The AXA motif, with small side chains at positions minus 1 and minus 3 relative to the cleavage point, is the most reliable structural indicator.
Cleavage site identification is the most technically demanding part of manual sequence analysis. The enzyme signal peptidase is specific in its requirements, and predicting exactly where it will cut requires accounting for multiple sequence positions simultaneously. This is where computational approaches become necessary for reliable results.
Manual inspection of short sequences is feasible. At scale, or when dealing with novel proteins where structural intuition is unreliable, prediction models are the standard approach.
The dominant models in signal peptide prediction use machine learning and artificial neural networks trained on curated data sets of known signal peptide sequences. The training process optimizes the model to distinguish signal peptides from other N-terminal sequences, including transmembrane helices and mitochondrial targeting sequences, which share some structural features.
SignalP is the most widely used tool in this category. Its versions have evolved from simple hidden Markov models to deep learning architectures, with each iteration improving accuracy and expanding the range of organisms covered. The current deep learning version processes input sequences and outputs a probability score for signal peptide presence, as well as a predicted cleavage site position.
The probability output is important context for interpretation. A high score reflects a strong match to patterns in the training data set, not certainty. The model's accuracy depends on how well its training data represents the protein in question. For proteins from organisms underrepresented in public databases, or for engineered fusion proteins, prediction confidence decreases.
Other prediction approaches include Phobius, which simultaneously models transmembrane topology and signal peptides to reduce false positives, and TMHMM-based pipelines used for membrane protein annotation. Each algorithm makes different assumptions and has different error profiles. Cross-referencing results across multiple models improves reliability.
Data mining of existing protein databases such as UniProt and the Protein Data Bank adds another layer of evidence. If a homologous protein with a characterized signal peptide exists in the database, alignment with the query protein can provide corroborating evidence for or against signal peptide presence.
Deep learning has substantially changed the precision ceiling for signal peptide prediction. Earlier models relied on fixed-window scoring or simple positional weight matrices. Contemporary architectures process entire sequences and learn long-range dependencies that earlier methods could not capture.
Convolutional neural networks were among the first deep learning architectures applied to this problem, followed by recurrent networks capable of modeling sequential data. More recently, transformer-based architectures have been adapted for biological sequence modeling. The logic parallels the way BERT, a language model originally designed for natural language processing, learns context-dependent representations by attending to all positions in a sequence simultaneously. Applied to amino acid sequences, transformer models treat each residue as a token and learn representations informed by the full sequence context rather than only local windows. This approach improves the model's ability to distinguish signal peptides from structurally similar but functionally distinct sequences.
Training these models requires large, carefully curated data sets with verified positive and negative examples. The quality of the training data determines the accuracy ceiling more than architecture choice alone. Benchmark studies comparing models consistently show that training data coverage across organism groups is the primary driver of generalization performance.
Accuracy and precision improvements from deep learning are most pronounced for proteins from less-studied organisms, where earlier models lacked sufficient training representation. For well-characterized mammalian proteins, performance differences between older and newer architectures are smaller.
The reliability of any signal peptide prediction depends as much on how scores are calculated and interpreted as on the underlying architecture. Understanding the data analysis layer helps researchers evaluate model outputs critically rather than treating probability scores as definitive answers.
Most prediction tools generate per-residue scores along the sequence, then aggregate these into a summary signal. A common aggregation approach applies a weighted arithmetic mean across positional scores, where positions near the predicted cleavage site receive higher weighting than distal positions. This concentrates the model's confidence signal around the most discriminating region of the sequence rather than averaging uniformly across residues that carry limited predictive information.
Correlation between structural features and prediction scores is an active area of methodological research. Studies have examined how well the hydrophobicity profile of the H-region correlates with model confidence scores, finding that in most architectures, the correlation is strong for canonical signal peptides but weakens for atypical sequences. This correlation analysis helps identify the classes of proteins where prediction tools are most and least reliable.
Measuring the efficacy of prediction algorithms requires standardized benchmarking data sets with verified ground-truth annotations. Commonly used metrics include sensitivity, specificity, the Matthews correlation coefficient, and area under the receiver operating characteristic curve. These metrics collectively capture both the detection rate for true signal peptides and the false positive rate for sequences that resemble signal peptides structurally but lack function.
Data set refinement continues to improve model performance. As new experimentally validated signal peptide sequences are published and deposited in databases, retraining cycles incorporate this evidence and progressively tighten the accuracy of prediction outputs.
The practical toolkit for signal peptide analysis consists of publicly available bioinformatics software, most of which is accessible through web interfaces or programmable APIs for integration into larger analysis pipelines.
SignalP, hosted by the Technical University of Denmark, remains the reference tool. It accepts FASTA-format input and returns position-specific scores, cleavage site predictions, and organism-group classifications. For large-scale analysis, the command-line version integrates with distributed computing environments and high availability workflows, making it suitable for proteome-wide annotation projects.
DeepSig and SPEPlip represent newer deep learning implementations that offer competitive accuracy, particularly for eukaryotic proteins.
For developers building custom analysis pipelines, Biopython provides programmatic access to sequence parsing and integration with database APIs. Many researchers build these pipelines within integrated development environments that provide code completion, debugging tools, and version control integration, which meaningfully reduces development time for complex analysis workflows. The choice between point-and-click web interfaces and programmatic tools in an IDE typically depends on scale: web interfaces suit single-protein queries and exploratory analysis, while programmatic pipelines are necessary for proteome-wide annotation or automated retraining workflows.
Database integration is a consistent requirement across all serious prediction workflows. Linking prediction output to structured databases enables downstream analysis of signal peptide conservation, evolutionary patterns, and disease-associated variants.
Computational prediction identifies candidates. Experimental validation confirms them.
DNA sequencing is typically the starting point. Cloning and expressing a protein of interest requires accurate sequence data, and any mutations in the signal peptide region will affect experimental outcomes.
Protein purification followed by N-terminal sequencing directly identifies the mature protein start site, confirming where cleavage occurred. This is direct biochemical evidence of signal peptide function.
Western blot is a foundational tool in signal peptide validation workflows. By running cell lysate and conditioned media fractions on a gel and probing with an antibody against the protein of interest, researchers can directly compare the size of the precursor form, which retains the signal peptide, against the processed mature form. A size shift between the intracellular and secreted fractions is direct evidence of signal peptide cleavage. Western blot is also used to confirm that signal-peptide-deleted mutants fail to secrete, providing the negative control data that validates the interpretation.
Mass spectrometry has become the dominant tool for characterizing signal peptide cleavage at the residue level. By analyzing peptide fragments from digested proteins, researchers can map the precise N-terminus of the mature form and confirm the predicted cleavage site.
Exosome analysis represents an emerging context for signal peptide research. Exosomes are small extracellular vesicles secreted by cells that carry a cargo of proteins, and the targeting of specific proteins into exosomes involves signal-mediated sorting processes. Characterizing which proteins are enriched in the exosome fraction relative to bulk secreted media helps researchers understand how signal peptide identity influences downstream trafficking decisions beyond the initial translocation step.
Flow cytometry has become increasingly important for quantitative signal peptide validation. When a protein of interest is tagged with a fluorescent marker and expressed in a cell culture model, flow cytometry enables single-cell measurement of surface or secreted protein levels across large cell populations. Comparing wild-type signal peptide constructs against mutant versions using flow cytometry gives a direct quantitative readout of trafficking efficiency.
Chromatography techniques are used in protein purification workflows to isolate secreted or membrane-associated proteins. Transfection of expression constructs enables functional comparison studies.
Reproducibility standards require that experimental results be formally reported with sufficient methodological detail for independent replication. Published studies reference their data sets and analytical methods with Digital Object Identifier (DOI) links, enabling accurate literature tracing. The use of well-characterized model organisms, including mouse models, provides a bridge between cell culture experiments and in vivo validation of signal peptide function in living systems.
Beyond the standard validation toolkit, several approaches extend the resolution and scope of signal peptide characterization.
Imaging-based methods contribute to localization studies. While CT scan technology operates at a resolution too coarse to visualize molecular trafficking directly, it is used in research contexts where protein localization is studied at the tissue and organ level in animal models, providing spatial context for secretion phenotypes observed at the cellular level.
Intramuscular injection of expression constructs is used in some in vivo validation studies, particularly in vaccine research where viral vector delivery of antigen-encoding sequences requires functional signal peptides for correct antigen processing and surface display. This delivery method provides a contrast with the cell culture models used for initial validation: where cell culture isolates the signal peptide function under controlled conditions, in vivo injection studies reveal how signal peptide efficiency translates to whole-organism protein expression contexts.
Mouse models remain the dominant in vivo system for signal peptide functional studies. Transgenic and knockout approaches allow researchers to assess how signal peptide mutations affect protein secretion and localization at the tissue level, with measurable downstream effects on physiology.
Signal peptide function is governed by physical and chemical principles that operate at the molecular scale. Understanding this layer clarifies why sequence features predict function as reliably as they do.
The biophysics of protein transport through the translocon channel has been studied extensively using single-molecule and reconstitution approaches. The translocon is not a passive pore; it actively facilitates insertion and assists with the lateral release of transmembrane segments into the lipid bilayer. The energetics of this process depend on the hydrophobic core of the signal peptide providing sufficient driving force for membrane insertion, while the flanking regions position the sequence correctly for recognition and cleavage.
Catalysis by signal peptidase proceeds through a serine-lysine dyad mechanism, distinct from the more common serine-histidine-aspartate catalytic triad of other serine proteases. This biochemical specificity is part of what makes signal peptidase a selective enzyme: it cleaves at the C-region motif and does not act on arbitrary hydrophobic sequences. Understanding this catalytic mechanism informs both prediction model design and inhibitor development.
Protein motility within the cellular environment, including lateral diffusion in the membrane and directed transport along cytoskeletal elements, depends on correct initial targeting by the signal peptide. Proteins that are mistargeted due to signal peptide dysfunction do not simply end up in a random location; they often enter degradation pathways, producing a measurable reduction in steady-state protein levels. The correlation between signal peptide integrity and downstream protein abundance is well documented in the experimental literature and forms the quantitative basis for functional validation assays.
In the human body, signal peptides are active across virtually every tissue and organ system. The liver is a particularly high-output organ for signal peptide-dependent secretion: hepatocytes synthesize and secrete the majority of circulating plasma proteins, including albumin, clotting factors, and complement components, all of which require functional signal peptides for entry into the secretory pathway and release into the circulatory system.
Red blood cell precursors in the bone marrow also depend on signal peptide-directed trafficking for the production of surface proteins, though mature red blood cells, which lack ribosomes, do not synthesize new proteins. The proteins that govern red blood cell identity and function are produced during differentiation, with signal peptides directing membrane proteins to their surface locations before the nucleus is expelled.
Lymphocytes, the immune cells responsible for adaptive immunity, depend on signal peptide-directed secretion for antibody production. B cells produce immunoglobulins in large quantities, and the signal peptides on both heavy and light chains are required for correct entry into the secretory pathway, assembly in the endoplasmic reticulum, and eventual secretion as functional antibody molecules.
Signal peptide dysfunction is connected to systemic disease through multiple mechanisms. In cancer, aberrant signal peptide sequences or mutations affecting signal peptide cleavage can result in the mislocalization of growth factor receptors and secreted ligands, contributing to dysregulated cell signaling. The connection between signal peptide biology and cancer is an active research area, particularly in the context of identifying tumor-associated secreted proteins as biomarker candidates.
Viruses exploit signal peptide biology at multiple levels. Viral envelope proteins use signal peptides to enter the host cell's endoplasmic reticulum, where they are glycosylated before transport to the cell surface for viral assembly. Viral vectors used in gene therapy and vaccine delivery encode antigen sequences with optimized signal peptides to maximize surface expression and immunogenicity. The signal peptide sequence in these viral vector constructs is often engineered specifically for the target cell type, reflecting how thoroughly signal peptide function has been integrated into applied biotechnology.
Signal peptides are conserved in function but variable in sequence across organisms. The same three-region architecture appears in bacteria, archaea, and eukaryotes, but with meaningful differences in both sequence and the machinery that processes them.
In bacteria, signal peptides direct proteins across the plasma membrane via the Sec or Tat translocon systems. Bacterial signal peptides tend to be shorter and follow tighter sequence constraints than their eukaryotic counterparts.
Archaea occupy a distinct branch in this comparison. Archaeal signal peptides resemble bacterial ones structurally, particularly in the hydrophobic core, but are processed by different signal peptidases. Type IV pilin-like signal peptides found in many archaea have a distinctive positively charged N-region and a different cleavage motif than either bacteria or eukaryotes. This distinction is relevant for researchers working with archaeal expression systems or studying extremophile proteins, where prediction models trained primarily on bacterial and eukaryotic data may underperform.
In mammals, signal peptides tend to be longer and more variable than bacterial or archaeal equivalents, reflecting the greater complexity of the eukaryotic secretory pathway. Evolutionary analysis of signal peptide sequences across these three domains reveals that the hydrophobic core is under the strongest selective constraint, while the flanking regions tolerate considerably more variation.
Signal peptides are not isolated molecular features. They sit within the broader context of protein synthesis and cellular physiology, with consequences that extend to tissue-level biology.
The secretory pathway, which signal peptides initiate, is responsible for producing the full complement of extracellular and membrane-bound proteins that tissues require for structural integrity and intercellular communication. This includes growth factors, extracellular matrix components, and receptor ligands that regulate processes such as cell proliferation and tissue remodeling.
In the context of skeletal muscle physiology, hypertrophy, the increase in muscle cell size driven by elevated protein synthesis rates, depends in part on the correct secretion and presentation of signaling molecules and matrix proteins. Signal peptides on secreted growth factors and collagen precursors ensure these molecules reach the extracellular environment where they act. Disruptions in signal peptide function in this context can affect the efficiency of the secretory pathway during periods of elevated anabolic demand, though this connection remains an area of ongoing research rather than established mechanism.
Metabolism more broadly depends on correctly localized enzymes and transporters, many of which reach their functional locations via signal peptide-directed transport. The endogeny of signal peptide sequences, meaning their origin as intrinsic components of naturally occurring proteins rather than engineered additions, is relevant to this context. Endogenous signal peptides have been shaped by evolutionary pressure to function reliably within the native cellular environment. Engineered or heterologous signal peptides introduced in recombinant protein production do not always replicate this reliability, which is why signal peptide optimization remains a practical concern in biotechnology workflows.
The scientific literature on signal peptides is extensive, distributed across molecular biology, structural biology, computational biology, and applied biotechnology journals. Navigating this literature effectively requires understanding how citation networks and database cross-referencing work in practice.
Citation in signal peptide research serves multiple functions beyond attribution. Citing the original publications that characterized a signal peptide provides traceability for experimental claims. Citing the prediction tool version and training data used in a computational analysis allows independent reproduction of results. These practices are part of the methodological transparency that reproducibility standards require.
Digital Object Identifier (DOI) links are the standard mechanism for stable reference to published work. When a signal peptide prediction result is reported in a publication, linking to the source database entries and tool documentation via DOI ensures that readers can access the same resources and verify the analysis. The adoption of DOI-based referencing across molecular biology has substantially improved the traceability of computational claims over the past decade.
Measuring the efficacy of prediction tools requires not just internal benchmarks but independent validation on held-out data sets that were not used in training. Published tool comparisons that use standardized held-out data sets provide the most reliable guidance for researchers selecting tools for their own workflows.
Signal peptide research is distributed across a global network of institutions, with concentration in regions with strong molecular biology and biotechnology infrastructure.
In the United States, academic medical centers and biotechnology companies have produced a substantial proportion of the foundational work in signal peptide biology and prediction tool development. The Boston and Cambridge corridor in Massachusetts is a particular concentration point, where institutions such as MIT, Harvard, and the Broad Institute, alongside a dense cluster of biopharmaceutical companies, have contributed to both basic research and applied development.
China has emerged as a significant contributor to peptide and protein research over the past two decades. Investment in life sciences infrastructure, including sequencing capacity and bioinformatics development, has supported a growing output of peer-reviewed work in signal peptide prediction and protein secretion biology.
Internationally, the European Molecular Biology Laboratory and its associated networks have played a central role in the development and maintenance of the databases and bioinformatics tools that underpin the field.
Scientific figures are a primary communication medium in signal peptide research. A well-constructed fig depicting the three-region structure of a signal peptide, with the N-region, H-region, and C-region labeled against a sequence backdrop, conveys information that would require several paragraphs of text to approximate in prose.
Standard visualization approaches in published research include linear sequence diagrams with shaded regions indicating structural domains, helical wheel projections of the H-region to illustrate the hydrophobic face, and schematic cross-sections of the endoplasmic reticulum membrane showing the translocation event.
In computational contexts, figures representing model performance typically show receiver operating characteristic curves, confusion matrices, and precision-recall plots. Data visualization also extends to database browser interfaces, where signal peptide annotations are displayed alongside full-length protein records. Understanding these visual conventions is part of the practical skill set for working effectively with sequence databases.
The path from signal peptide discovery to commercial product involves multiple translational steps, each of which depends on the foundational identification and validation work described throughout this post.
In drug development, signal peptide optimization is part of the manufacturing process for biologic therapeutics. The adoption of signal peptide engineering as a standard practice in biologics workflows reflects how thoroughly the field has internalized the impact of signal peptide choice on product yield and quality. A signal peptide that performs well in one expression system may reduce titer significantly in another, which is why expression system-specific optimization remains a routine step rather than a solved problem.
The biotechnology product pipeline from bench to commercial scale requires signal peptide characterization at multiple stages: initial expression construct design, cell line development, scale-up, and regulatory submission. At each stage, documented evidence of signal peptide function and cleavage contributes to the analytical characterization package that regulatory agencies review. This positions signal peptide identification not just as a research exercise but as a component of the quality framework governing biological product development.
Signal peptide engineering is a productive area of applied biotechnology. In recombinant protein production, the choice of signal peptide affects secretion efficiency substantially. Optimizing the signal peptide sequence for a given expression system, whether bacterial, yeast, insect cell, or mammalian, can increase yields without changing the mature protein sequence.
Monoclonal antibody production depends on functional signal peptide sequences for heavy and light chain secretion. Signal peptide optimization is a routine step in biologics manufacturing workflows.
Emerging applications include the use of signal peptide prediction as a component of proteomics annotation pipelines. Large-scale research and development workflows that process entire proteomes require automated, accurate signal peptide classification to distinguish secreted proteins from cytoplasmic and membrane-integral proteins. This classification step feeds into downstream analyses ranging from biomarker discovery to systems biology modeling.
Signal peptides are one layer of a much larger picture of how peptides function at the molecular level. If you are building a research framework around peptide biology, the Project Biohacking Peptide Calculator is a practical starting point for working with peptide parameters in a structured way. For sourcing decisions, theVendor Directory covers quality signals and third-party verification standards relevant to research-grade procurement.
Signal peptides are identified by analyzing amino acid sequences for a hydrophobic central region, a positively charged N-terminus, and a cleavage site in the C-region following characteristic structural patterns.
Machine learning models, artificial neural networks, and deep learning architectures including transformer-based models trained on curated sequence data sets are the primary tools for signal peptide prediction, with SignalP being the most widely used.
Mass spectrometry, western blot, N-terminal protein sequencing, flow cytometry-based trafficking assays, exosome fraction analysis, and chromatography-based secretion assays are used to validate computationally predicted signal peptides.
Yes, signal peptides differ across bacteria, archaea, and eukaryotes in sequence length, cleavage motif, and the translocon machinery they engage, with mammalian signal peptides generally longer and more variable than prokaryotic equivalents.
Bacteria use signal peptides to secrete virulence factors, and viruses use them to direct envelope proteins to the endoplasmic reticulum for processing, with viral vectors in gene therapy and vaccines engineered to optimize signal peptide function for surface antigen display.
It determines protein localization, supports accurate annotation of secreted and membrane-targeted proteins, and informs biotechnology applications including recombinant protein production and drug development.

About the Author:
Jeff Nunn is the founder of Project Biohacking. With over 30 years of biohacking practice, he applies decades of self-experimentation methodology to peptide research, dosing math, and vendor evaluation.
Important Disclaimer: The content on Project Biohacking is for educational and informational purposes only and is not intended as medical advice, diagnosis, or treatment. Always consult a qualified healthcare professional before making any changes to your health regimen, starting new supplements, peptides, or protocols. Nothing on this site establishes a doctor–patient relationship, and you use the information at your own risk. Research compounds discussed here are sold for laboratory research purposes only and are not approved for human or veterinary use or consumption.
“For educational use only. Not medical advice. Read our full disclaimer.”
+1 214-278-4039
All Rights Reserved | Project Biohacking